

Advanced, all-in-one, cloud-based clinical metadata repositories are helping to standardise clinical trial design and build, increase throughput, and decrease costs
This article is taken from International Clinical Trials August 2021, pages 22-24. © Samedan Ltd [digital edition available here]
The clinical trial landscape is changing. More methods and technologies to streamline and fast-track drug approval and commercialisation are becoming available. As such, there is growing pressure to get more studies through the pipeline faster, while continuing to ensure patient safety. Organisations are constantly looking to identify and improve the elements of designing and building clinical trials that cause the biggest time and cost inefficiencies. One major hurdle is the methods currently being used by many organisations to collect, manage, and analyse the vast amounts of clinical trial metadata, as they don’t allow for standardisation.
Historically, many pharmaceutical, biotechnology, and CRO facilities have collected clinical metadata using multiple desktop applications, which need to be installed and maintained manually, resulting in siloed repositories that are difficult to manage. Other organisations have used various in-house or third-party systems. Stakeholders, such as data managers, biostatisticians, and clinicians, therefore, end up working on their own parts of the process, holding metadata in different formats across several disparate systems in multiple locations. This makes standardisation of metadata extremely challenging, and some would say impossible to achieve.
The Clinical Data Interchange Standards Consortium (CDISC) and the FDA have worked closely together to make processing and reviewing clinical trials more effective by regulating metadata standardisation.
Different stakeholders within organisations building clinical trials are key to ensuring regulatory compliance. Biostatisticians, for example, are responsible for producing high-quality, compliant datasets, such as study data tabulation models (SDTMs), for submission to regulatory bodies, while also ensuring metadata is presented in a format that can be easily understood by clinicians to drive well-informed decisions. Data managers, on the other hand, are tasked with ensuring clinical trial data are collected, managed, and reported in a clear and accurate manner. However, standardising metadata in the required formats can be challenging when different stakeholders use multiple disparate systems within an organisation. In particular, this is the case when organisations with different protocols and technologies merge.
|
To answer these demands, advanced, all-in-one, cloud-based clinical metadata repositories (CMDRs) and automation platform technologies are being introduced to help organisations quickly build clinical trials in compliance with regulatory standards, such as those set by CDISC. These systems store metadata in a single, centralised repository, which removes the problems associated with siloed metadata. As a result, metadata accuracy, consistency, and quality are considerably improved. This increases clinical trial throughput, decreases costs, and, ultimately, brings life-changing treatments to patients more quickly.
|
Streamlining Clinical Trials With CDISC Standards
Compliance with CDISC standards can be challenging. Organisations must understand and comply with complex requirements. Moreover, stakeholders must constantly update their internal systems as CDISC regulations change, often by attending training sessions and conferences, and regularly reviewing CDISC and FDA websites. As such, some companies have been approaching CDISC compliance as an afterthought – something to work on at the end of the trial just before submission for regulatory approval. This approach is counterproductive, and can cause major delays at the end of the process. Instead, building CDISC standards into the earliest planning and design stages of clinical trials is a crucial way to expedite submissions and increase the chance of success.
The CDISC standards model covers planning through to data analysis before submission of metadata to regulators. Data tabulation and analysis processes are regulatory requirements in the US and other countries (Figure 1, see red box). Industry standards such as SDTM, analysis data model (ADaM), and define-extensible markup language (Define-XML) are built to ensure regulators receive studies in a consistent format every time, helping to streamline the review process (Figure 1, see red box). This whole process ensures that metadata can be mapped back to case report form (CRF) data, and it defines what should be included in the analysis datasets. This all has to be described in the correct format (Define-XML), and must also align with National Cancer Institute (NCI)-controlled terminology standards.
Planning and data collection stages are not governed by regulatory requirements, but are an integral part of the internal processes of organisations when building clinical trials. These stages help to integrate systems and produce CRFs that are consistent with the required downstream standards, while making sure the necessary data will be collected and available in the appropriate format. While compliance with processes like those described by the Operational Data Model (ODM) and Clinical Data Acquisition Standards Harmonization are not required for regulatory submissions, connecting them with data tabulation and analysis in a standardised way greatly facilitates metadata management in accordance with regulatory requirements.
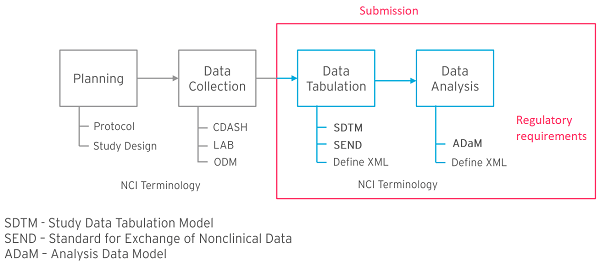 Figure 1
Figure 1
Metadata Standardisation Technologies: The Future of Clinical Trial Design and Build
Automated technologies that support organisations to standardise their clinical metadata management and ensure regulatory compliance are becoming increasingly available. Ideally, technologies should cover the entire process of clinical trial design and build. However, some platforms aiming to facilitate standardisation are limited in scope, focusing purely on defining standards, but not going far enough to generate datasets ready for submission to regulatory bodies.
Modern, all-in-one, cloud-based CMDR and automation systems with in-built CDISC compliance are now available, delivering time and cost efficiencies throughout the entire process, from setup and analysis right through to submission. Crucially, these CDISC-driven platforms define processes and data terms in a consistent manner that regulators have agreed upon.
The benefits of using MDR systems include standardising clinical metadata to achieve CDISC compliance through all stages of clinical trials. This encompasses planning to analysis, including industry standards, such as SDTM and ADaM, while all being generated with NCI-controlled terminology. Moreover, standardised metadata can be reused as and when needed, eliminating the need to create new content from scratch when looking to set up a new trial. CRFs, for example, can be reused across any leading electronic data capture system. As a result, study build is accelerated and resource investment is minimised.
Advanced CMDR systems also automatically stay up to date with changing CDISC standards through software updates and plugins. These features help to reduce reliance on individuals to understand complex regulatory requirements and manually keep track of changes. When importing existing metadata into a CMDR platform, a validation check function alerts users to any issues or non-compliance, prompting corrective action. Likewise, the latest CMDR systems use CDISC-compliant templates to allow for the creation of new metadata, guaranteeing compliance. Furthermore, if users introduce any edits to the metadata that stray beyond the boundaries of CDISC compliance, the platform alerts them so that they can address the issues.

Deploying CMDR systems may be seen as a major investment for organisations, requiring training of large numbers of users, and incurring high costs. Initially, a considerable amount of time and effort must be invested in order to reap the long-term, ongoing benefits. However, by adopting an effective long-term strategy, organisations can draw significant return on investment from metadata reuse and data management automation, even after just a handful of trials.
Conclusion
Standardising clinical study design and build can improve an organisation’s cost effectiveness, efficiency, and quality, and streamline internal processes. All-in-one, cloud-based, CDISC-compliant CMDRs and study automation platforms support this, standardising metadata from study planning to data acquisition, tabulation, and analysis. This allows content to be reused, and enables real-time data visibility.
Implementing MDR adoption throughout the industry can be challenging, and systems must demonstrate real value to organisations. However, once fully integrated into the organisational structure they can reshape an organisation’s capacity to enable better quality, faster trials, thereby expediting the whole drug development process.


