

Expediting Clinical Trials with Cloud-based Metadata Repository and Study Automation Technologies
This article is taken from Journal for Clinical Studies Volume 13 Issue 4, "Expediting Clinical Trials with Cloud-based Metadata Repository and Study Automation Technologies" .
|
The COVID-19 pandemic put clinical trials firmly in the spotlight. Powered by the urgent need to combat this global threat, clinical trials have been created and conducted at an astonishing rate. At the time of writing, over 9,400 clinical trials had been submitted to the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) [1]. By challenging the traditional view on the structure and format of clinical trials and applying the Clinical Data Interchange Standards Consortium (CDISC) requirements, pharmaceutical, biotechnology and contract research organisations (CROs) have expedited trial timescales. Study automation platforms and next-generation, cloud-based clinical metadata repositories (CMDRs) have been at the centre of these changes, standardising and reusing metadata and automating manual and error-prone study builds.
|
The learnings from the COVID-19 response are reinforcing the need for strategic change in clinical trial design and the generation of submissible metadata for regulatory approval. This article discusses how study automation platforms and all-in-one, cloud-based CMDRs can help organisations start trials quicker, deliver rapid insights and streamline regulatory approval.
The Need for Speed in Clinical Trials
The success of a pharmaceutical or biotechnology organisation rests on its ability to bring new therapies to market, and this, in turn, puts mounting pressure on businesses to push clinical studies through the pipeline faster and more cost-effectively. For contract research organisations (CROs), efficiency is everything and time is literally money; yet, for all companies, this focus on speed must not come at the cost of patient safety or robust efficacy evidence.
For too long, fragmented internal processes, legacy systems and metadata siloes have created bottlenecks in clinical trial design and execution. Manual workflows and disconnected worksheets introduce transcription errors and repetitive work leads to delays.
When metadata rules are reinvented for each project, the risk of inconsistencies increases, and trials don’t amass the knowledge or successes of previous trials. This lack of standardisation means extensive and expensive metadata rework before regulatory submission, adding further delays and costs.
Although there are many technologies available to alleviate these problems, they are limited in scope and often aimed at individual issues, such as standards management, study design or data conversion automation. When solutions operate in siloes, disconnects restrict returns and organisations, overwhelmed by the potential risks, choose the path of inactivity. Only a holistic approach to metadata management and automated trial design can deliver the benefits needed to drive strategic change and maximise returns throughout the clinical trial process.
Viewing Standardisation as a Strategic Tool
Since regulatory approval is reliant on the diligent evaluation of data, regulatory bodies, such as the Food and Drug Administration (FDA), stipulate data standardisation requirements in order to receive, process, review and archive submissions effectively. To begin to address metadata inconsistencies, CDISC [2] was created, in collaboration with the FDA, to drive interoperability through data standardisation and accelerate the design, build, analysis and submission of clinical trials (see Figure 1).
The items within the data tabulation and data analysis sections are regulatory requirements and refer to the standardisation of the structure and format of submissible data, including:
- Standard for Exchange of Non-clinical Data (SEND) – the
content and structure of non-clinical data collected in a clinical
trial, structured for submission. - Study Data Tabulation Model (SDTM) – the content and structure of the data collected in a clinical trial, structured for submission.
- Analysis Data Model (ADaM) – the content and structure of the analysis datasets generated from SDTM data.
- Define XML – the metadata format used to describe the content and structure of the SEND, SDTM and ADaM datasets.
- NCI terminology [3] – the consistent language used to make all terms in collected data clear.
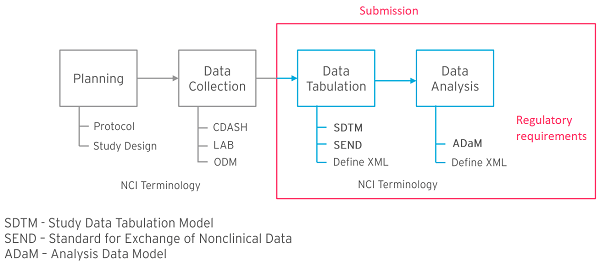 Figure 1: CDISC standards model
Figure 1: CDISC standards model
CDISC standards, implemented in the planning and data collection stages, are not regulatory requirements. Rather they’re integrative tools, defining the studies in common terms and creating the case report forms (CRFs) that are used to collect standardised data. Too often, organisations focus on the regulatory elements of CDISC, like a compliance afterthought or a tick-box exercise, rather than seeing it as a strategic tool to streamline the process and deliver efficiencies. By not incorporating CDISC standards into the process, compliance can cause considerable costs and delays as data managers rework data to meet the standards. Put simply, organisations feel the pain of compliance without reaping the rewards. When CDISC is viewed as the strategic tool that it was intended to be, clinical data teams can refocus trials, maximise efficiencies by optimising system integration, and create pre-approved data collection instruments that deliver automated, standardised data.
By automating the creation of submissible data, early safety views can be taken and in-trial adjustments made, allowing trials to become more flexible and adaptive. Standardisation becomes the central control, enabling organisations to focus on the real purpose of clinical trials: to bring life-saving therapies to people who need them in a faster and more cost-effective way.
The Power of All-in-one, Cloud-based CMDRs
Although standards governance has been evolving over the past two decades, organisations have only recently started to see its value. And while technology has also evolved to keep pace, adoption is in its infancy. But attitudes are changing. If organisations are ready to transform and embrace standardisation as a tool for profound and long-lasting strategic change, then CMDRs and study automation technologies will become essential for effective and efficient clinical trial design, collection and analysis.
An MDR creates a central hub for clinical trial metadata. It is a core, single-source-of-truth where forms, annotations, terminologies, datasets and much more are stored and can be accessed, anytime and anywhere by all who have the necessary permissions. Firstly, this secure bank allows visibility of all historic and current metadata, providing the information needed to build and conduct clinical trials in the most automated way, reusing content and decreasing the time and effort needed. Secondly, by having standards built-in and updated as new versions are released, the MDR ensures all stakeholders have access to compliant metadata that meets the needs of the current governance and requirements of regulatory bodies. Finally, the MDR provides a place for collaboration and controlled
change management for future studies (see Figure 2).
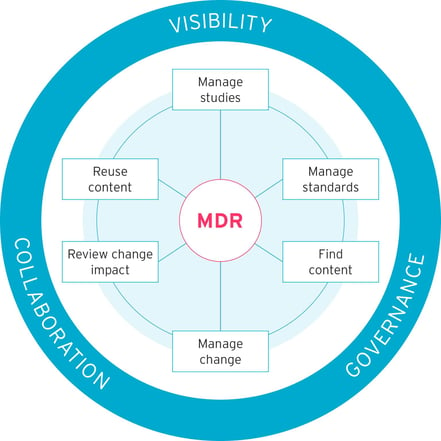 Figure 2: An MDR system facilitates visibility, governance and collaboration
Figure 2: An MDR system facilitates visibility, governance and collaboration
MDRs have advanced with standardisation and can now deliver a holistic approach to metadata storage, control and automated trial design. A robust and effective CMDR will be:
- Fully integrated – built as an all-in-one software suite, removing
the previously siloed approach to metadata construction, storage
and transfer. - Cloud-based – with no need for server space, infrastructure or inconvenient upgrade cycles. Cloud-based systems are accessible from any location and can be built to give a personalised view based on user permissions.
- Control-based – an MDR must be the single-source-of-truth.
Changes can only be made if they have been submitted and approved. - Standardised – built-in templates, assets and data are automatically created in accordance with CDISC standards, with no rework.
- Configurable – to accelerate the construction of eCRFs and maximise the reuse of metadata, MDRs must be shaped to meet organisational structure and workflows. They can also be configured to generate existing specification formats, reducing the amount of retraining needed.
- Compliant – MDRs should be updated when new standards are released. Proposed changes should be mapped, showing owners the impact of new versions and facilitating migration to them.
- Analytical – when a metadata change is requested, its impact should be modelled across the system, allowing for subjective and evidence-based decision-making.
- Connectivity-driven – plug-ins can join the MDR to external
systems to exchange data instantly.
MDRs Provide the Basis for Traceability, Consistency and Compliance
By integrating CDISC standards and installing an all-in-one, cloud-based CMDR, organisations can maximise strategic empowerment. Improving the consistency, storage and compliance of metadata leads to quality enhancements and increased efficiencies within the clinical trial process. When reuse increases, processes become more streamlined and trial design and execution time decreases.
High-quality, accurate metadata can become the basis for fast, right-first-time eCRF design before electronic data capture (EDC) system creation. The eCRF can be viewed and interrogated before the time-intensive EDC creation takes place. Some MDRs will allow EDC creation directly from the study specification, which further reduces the design time by removing manual builds and transcription errors.
Investing in a cloud-based CMDR that enables eCRF creation for multiple EDCs further improves its usefulness and accommodates the nuances of differing trials.
MDRs that combine study design automation with standardised data collection and analysis maximise their usefulness and return on investment (ROI). When standardised data is specified during trial design stages, it is created in a ready-to-submit format, allowing early views that can highlight any issues. Subsequently, with prompt in-trial adjustment and early regulatory submission, drug development timescales can be dramatically reduced. By standardising metadata and automating trial builds and generation of submissible data, all-in-one, cloud-based CMDRs can deliver faster, more accurate and more cost-effective trials.
Making Changes Now for the Long Term
Despite the clear benefits offered by all-in-one, cloud-based CMDRs, the adoption of this technology has been lagging. Whether small or large, pharmaceutical, biotechnology and contract research organisations often evolve organically, absorbing expertise, workflows and even other businesses as they move through a changing medical landscape. The task of assimilating large, siloed datasets and manual processes is not to be underestimated, and the complexity of legacy systems can certainly add to the burden. However, the short-term benefits of inertia are quickly outweighed by the burden of continuing to use manually intensive and error-prone workflows. As clinical trials increase in number and as organisations respond to the needs of a growing and ageing population, agility, speed, and cost will be key success factors in study design and operation, directly correlated to the ability of an organisation to increase its suite of marketable therapies.
Next-generation MDRs have certainly accommodated the valid concerns raised around integration, and have been designed to minimise the effort needed. Some MDRs offer seamless integration with existing platforms, and many feature application programming interfaces (APIs) and plug-ins to provide safe and secure data flow (see Figure 3).
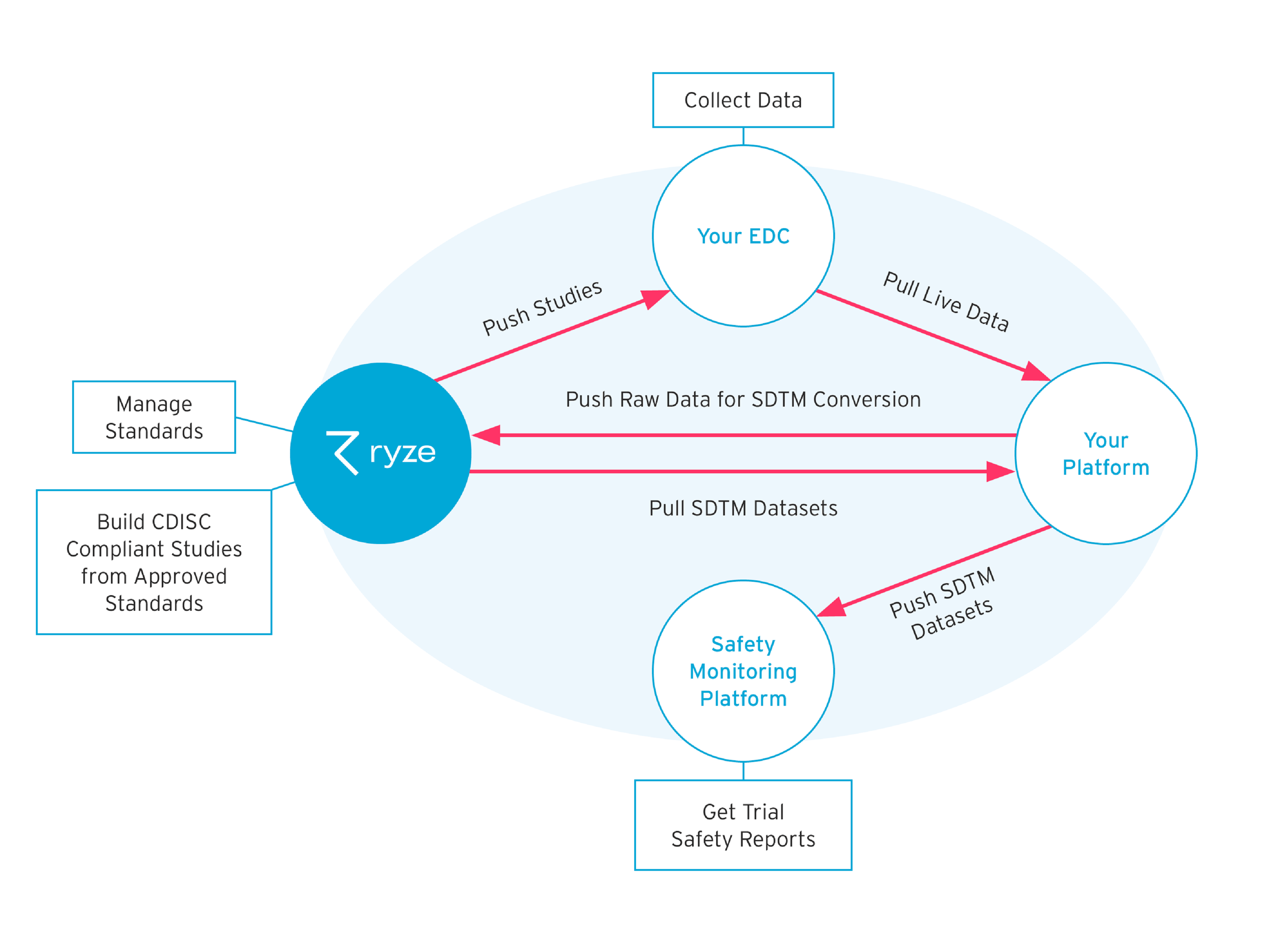 Figure 3: The integration of all-in-one, cloud-based clinical MDRs with existing technology through the use of APIs
Figure 3: The integration of all-in-one, cloud-based clinical MDRs with existing technology through the use of APIs
The key driver behind MDR integration is to build a space for users to create, view and consume metadata. When MDRs are integrated with existing technology, they can be brought online quickly with minimal user training; with a staged approach to integration, stakeholders are aligned. Each benefit, such as data standardisation or EDC build integration, can be fully realised before moving to the next. With a focus on usability, MDRs can quickly realise their full potential and deliver fast ROI, sometimes within just a couple of trials. This short-term investment generates long-term wins as MDRs and study automation technology evolve and grow with the business, further improving clinical trial efficiencies and directly impacting clinical trial successes and regulatory approval rates.
COVID-19 Vaccine Development: A Cloud-based CMDR Success Story
Although the ability to automate standardised data collection came before the COVID-19 pandemic, the need to develop therapies at an unprecedented speed brought its usefulness into the spotlight. All-in-one, cloud-based CMDRs and study automation platform technologies have been used in several high-profile COVID-19 studies. MDRs were used to create eCRFs during trial design, for rapid review and approval, while also simplifying and expediting the build of multiple EDCs directly from the study specification.
This process saw numerous studies get underway within weeks of vaccine development. However, in the race to reduce the typical 10-year development timeline, early indicators of trial success and accelerated approval were critical. Programmes began to release Phase I/II data as early as July and August 2020, and this was due in part to the rapid conversion of raw data to submissible SDTM files. The dynamic nature of these studies meant that metadata changes were frequent.
Many trials began to deliver daily data to sponsors, enabling an early safety and efficacy picture to emerge and facilitating expedited approval. This rolling review of trial progression relied heavily on standardised data; when this was incorporated at the heart of trial design, companies were able to gain rapid insights and adapt accordingly, dramatically reducing the time needed to bring vaccines to market.
Building All-in-one, Cloud-based CMDRs for the Future
All-in-one, cloud-based CMDRs provide the stable foundation to deliver the strategic change that drives faster, more accurate and more cost-effective clinical trials. MDRs were created to integrate siloed systems and allow disparate users to create, view and consume metadata. And APIs are the tools through which this happens. Platform developers are already working on the technology that will deliver the clinical trials of the future, using the considerable knowledge amassed in the past few years. Automation is already supported by using data from external systems to trigger SDTM conversions, and returning generated datasets to the original system. APIs in external systems can also be used to push or pull data between platforms, using live data from EDCs to automate SDTM conversions throughout the trial lifecycle. As third-party solutions extract insight and APIs continue to feed metadata and data, more control and automation can be generated through the MDR. The CDISC 360 project [4] is now looking to build on existing standards to drive end-to-end, metadata-driven automation and derive maximum ROI throughout studies.
Already, clinical trials are starting to make use of non-traditional, real-world metadata, such as that collected through wearable devices and patient-generated responses. As organisations make use of this real-world, less structured and semantically rich information, metadata standardisation will be ever more important for regulatory approval. As researchers extract real-world meaning from clinical trials, MDRs will be critical tools in the quest to structure, control, organise and extract these insights.
Investment in all-in-one, cloud-based CMDRs already brings immediate benefits: faster ROI, expedited trials, more cost-effective programmes and more accurate clinical data. But it is the wider and longer-lasting strategic benefits that will generate a competitive edge as organisations develop the clinical trials of the future. Investing in future-proofed technology that delivers both short-term and strategic benefits will ensure that organisations are prepared for the changes that will come.
REFERENCES
1. https://www.who.int/clinical-trials-registry-platform, visited on 30 April 2021.
2. https://www.cdisc.org/, visited on 2 May 2021.
3. https://ncit.nci.nih.gov/ncitbrowser/, visited on 30 April 2021.
4. https://www.cdisc.org/cdisc-360, visited on 2 May 2021.


