

This article is taken from international-biopharma.com, "Could Standardised Metadata be the Key to Optimising and Expediting Clinical Trials?" [digital edition available here].
|
Clinical trials still offer the most effective way of testing the safety and efficacy of new drug treatments, and as more drugs are brought through development each year, the number of trials conducted grows (see figure 1). Indeed, at the time of writing, there are currently 390,644 registered studies live in 291 countries. Although clinical studies are tried and tested, they are by no means static; trials are constantly evolving in response to patient needs and changing technologies, incorporating more complex and adaptive designs, and allowing for more detailed data collection. Together with the need to comply with more stringent regulations and frequently updated standards, clinical trial managers are under increasing pressure to deliver optimised and streamlined trials that deliver safe and efficacious drugs faster and more cost-effectively. Despite these pressures, many organisations still rely on manual processes, disparate systems and fragmented workflows to manage the metadata that supports and powers clinical trial design, data collection and regulatory submission. These traditional and out-of-date models can cause errors, inconsistencies and delays. Costs can soon escalate as trial managers struggle to find, use and manage metadata and, the issues don’t stop there, as when it comes to preparing metadata for regulatory submission, a longwinded programme of rework is often needed. This article explores how the use of next-generation, all-inone, cloud-based clinical metadata repositories (CMDRs) can provide the automation, standardisation and control needed to optimise clinical trials.
|
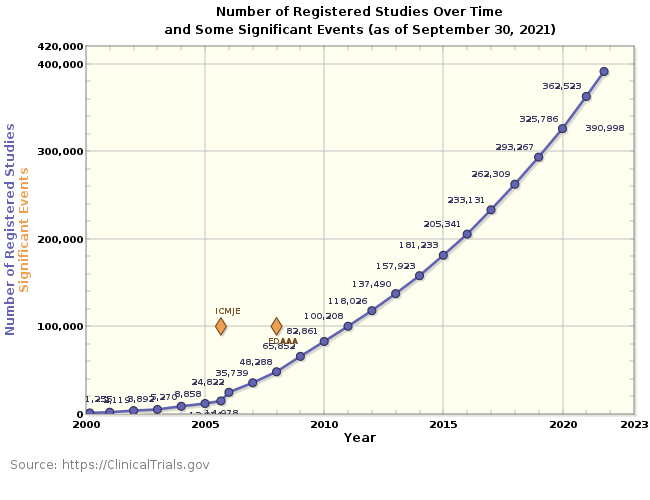
Figure 1 – Number of registered studies on the ClinicalTrials.gov website since its inception in 2000. In September 2005, the International Committee of Medical Journal Editors (ICMJE) started requiring trial registration as a condition of publication and in December 2007 registration was required in line with the Food and Drug Administration Amendments Act (FDAAA).
Metadata: The Underlying Framework for Successful Trial Design and Execution
Metadata underpins every clinical trial. It is the consistent framework that enables stakeholders to access, monitor, log and track data, and it is critical to every stage of a trial, from design to regulatory submission.
Effectively managing metadata is critical to delivering fast, efficient and cost-effective trials that contain robust and reliable data. But to optimise the use of metadata, team members need to have access to the correct versions, in a readable format, when they need it.
When you think of metadata, regulatory submissions often spring to mind and, indeed, it plays a critical part in regulatory review. In fact, the US Food and Drug Administration (FDA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA), require metadata to be submitted in a standardised format: that defined by the Clinical Data Interchange Standards Consortium (CDISC). This stipulation is to ensure that results can be easily and consistently understood and processed during regulatory review.
However, CDISC standards aren’t merely intended as a tick box for regulatory submission. In fact, they’re a strategic tool to streamline the design and build of clinical studies, reduce the effort needed to build a trial from scratch, and automatically generate submission-ready data and metadata. Yet, in practice, metadata is often manually reworked to comply with the standards at the end of a trial, consuming considerable time and resources, and potentially resulting in errors.
Despite the importance of easily accessible, accurate metadata to a clinical trial’s cost and success, many organisations don’t have the systems in place to maintain, control and access these valuable assets for all parts of a clinical trial’s lifecycle.
The Growing Challenge of Managing Metadata
The real challenges of managing metadata come from its legacy and the complex networks that organisations make as they expand. The real-world practicalities of maintaining and using metadata are, therefore, complex and fraught with issues:
- Locating Metadata is Difficult – it is often held in different
formats, folders and systems, and owned by different
stakeholders. - Managing Metadata in Unconnected Files Can Introduce Errors – siloes form and metadata can be lost as it is manually transferred, which can lead to transcription errors or deletion.
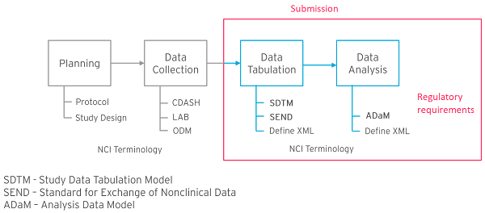
Figure 2 – CDISC standards model.
- Change Control is Inadequate – approvals may be managed through multiple email chains, making it hard to maintain accurate change history and version control.
- Impact Assessments are Lacking – when changes are requested, it’s difficult to see how they might impact other assets, standards or relationships with other metadata.
- Metadata isn’t Standardised – when the planning and data collection parts of CDISC standards are not adequately embedded, the data tabulation and analysis metadata outputs are not automatically standardised. Because these standards are required for FDA and PMDA submission, metadata is often manually reworked for compliance at the end of the trial (see figure 2), causing delays and the potential for errors.
- Metadata Can’t be Reused – it makes sense to reuse preapproved metadata, but without standardisation, version control and centralised storage, stakeholders can’t access this. Assets, such as electronic data capture (EDC) systems and electronic case report forms (eCRFs), need to be created afresh each time, often manually – a time-consuming process.
- A Trial’s Data Can’t be Validated Until Completion – this means that errors or quality issues might not be caught or addressed early. Cohort-level trends, such as poor responses or adverse effects may also be missed, which will limit trial adaptability.
- CDISC Standards Often Change – the constantly evolving nature of trials and their review means that standards must change. This means that experts are needed to keep abreast of developments and assess their impact.
Together, these issues mean that metadata isn’t carefully controlled, updated and accessible. In turn, this leads to the risk of quality issues creeping into trial designs and stifles the collaboration needed for a continuous improvement environment. Without collaboration and proper change control, changes could start to impact other projects and errors are more likely to occur.
CMDRs: Unleashing the Power of Metadata to Drive Effective Clinical Trials
The key to unlocking the full power of metadata, and its unique ability to optimise clinical trials, is to build a single-source-of-truth; a place where metadata can be centralised, standardised and controlled. The latest, all-in-one, cloud based CMDRs provide this hub, with the capability to hold the huge volume of study metadata that exists as organisations run multiple, evermore complex clinical trials (see figure 3).
With next-generation CMDRs, all stakeholders can access compliant, consistent and reusable metadata in a readable format, and monitor and track it for their specific purposes.
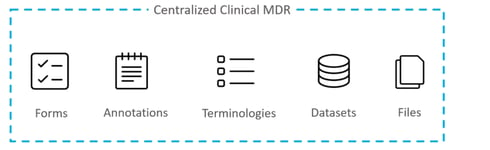
Figure 3 – The latest all-in-one, cloud-based CMDRs provide a central infrastructure for all metadata relevant to an organisation’s clinical trials.
This single-source-of-truth also creates a space for governance, visibility and collaboration (see figure 4), which drive effective and efficient trials. CMDRs provide this unique capability through automation, standardisation, change control, in-trial validation and integration with other trial software.
CMDRs Provide the Means for End-to-End Standardisation and In-Trial Validation
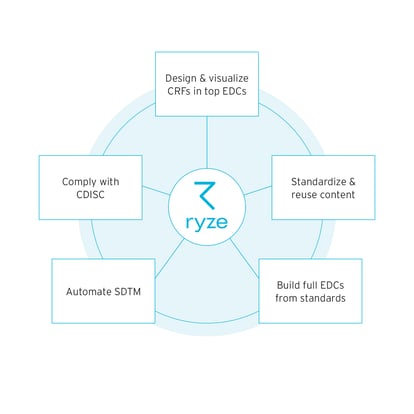
Figure 4 – CMDRs enable visibility, governance and collaboration in one central single-source-of-truth.
A critical attribute of next-generation CMDRs is that CDISC standards are built into their framework. Each stage of the CMDR is built with the next in mind, fostering seamless integration and inheritance between the stages.
Since studies are designed and built with pre-approved, compliant metadata, they deliver submission-ready outputs, with no rework needed. By cutting out the manual tasks of aggregating and reworking metadata at the end of a trial, timelines and the risk of errors are greatly reduced. Built-in CDISC standards offer another important function: templates can guide users to the correct versions for their submittable data, such as Study Data Tabulation Model (SDTM), Standard for Exchange of Nonclinical Data (SEND) and Analysis Data Model (ADaM).
Since validation is integral to a CMDR, any compliance deviations are automatically flagged by the system, preventing late-stage validation issues. Once CDISC-compliant metadata is approved, it can be stored and made readily accessible to other users. This pre-approved status forms the basis for the creation of organisational governing standards that stipulate how metadata is used.
Automation and Streamlining: The Driving Forces for Optimal Efficiency
Standardised, pre-approved and centralised metadata can be used to automate and streamline tasks, unleashing the full power of the CMDR to optimise trials and increase efficiency.
A manual and time-consuming task in the creation of clinical trials is the building of the electronic data capture (EDC) system. Sometimes trials will use multiple EDCs, which requires manual transcription into different software systems. Multiple EDCs can now be created directly from the trial brief within some of the latest CMDRs, saving a significant amount of time and reducing the errors that can arise from the manual translation of briefing files. Since the CMDR contains pre-approved CDISC-compliant metadata, the EDC is created to output compliant data, eliminating post-trial rework.
Some next-generation CMDRs can show electronic case report forms (eCRFs) before the more time-consuming EDC creation. This gives an early view of how a form will look, enabling review and approval to start sooner and progress quicker. Any underlying problems can be addressed early in the trial’s design, saving time later on when changes may take longer to implement.
Communication with EDC systems is just one function of the integration capability of some CMDRs. Using application programming interfaces (APIs), CMDRs can communicate with an array of trial software and safety programmes (see figure 5). APIs provide a simple and accurate way to push and pull metadata between the parent system (the CMDR) and auxiliary systems; manual processing and transfer of data are, therefore, reduced, while the CMDR continues to control the single-source-of-truth for all trial metadata.
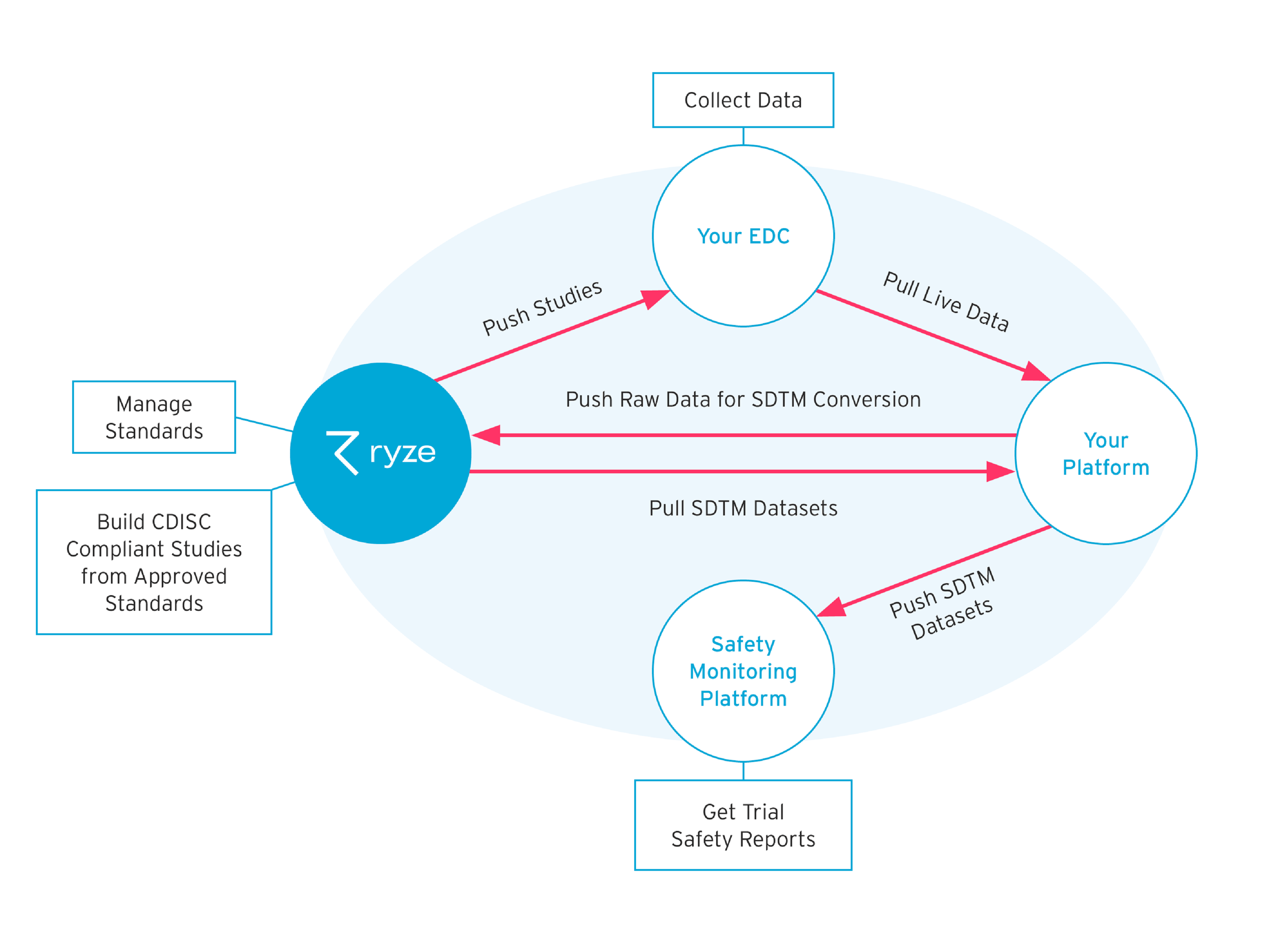
Figure 5 – Some CMDRs use APIs to transfer metadata and communicate with other Clinical trial software.
A key benefit of a CMDR is that it provides the ability to reuse metadata. Considerable effort goes into defining and validating metadata and reusing it saves time, especially when trials are complex, involve large cohorts or run concurrently.
Mappings, annotations, controlled terminology and datasets can be reused, meaning EDCs and eCRFs don’t need to be created afresh each time, and data quality and consistency are improved.
Checks, Audits and Change Control
A CMDR system will likely be accessed by multiple stakeholders all looking for metadata to inform their part of the clinical trial process. Lifecycle states must, therefore, be created for standards and studies to define how metadata is used and how it can be changed. When metadata is rejected or retired, then it must have checks in place to prevent its use in live studies.
CMDRs provide the structure to set these lifecycle states as well as the checks and barriers that stop non-approved metadata from making its way into a study.
This structure is only the start. CMDRs create a deep understanding of the relationships that exist in and around metadata, and this underpins a change control protocol.
If a change is requested, such as adding, editing or retiring metadata, its impact on related assets can be evaluated before the change is made. The relational framework with the CMDR makes change control possible and gives the ability to track metadata. In a CMDR, user interactions create an audit trail.
Whenever a user works with metadata, or requests or approves a change, this is documented and evidenced within the system. The automatically created trail further reduces the amount of work required to collate and validate metadata for regulatory submission.
CMDRs: Meeting the Multiple Challenges of the Modern Clinical Trial
In response to growing and unmet health needs, pharmaceutical, biotechnology and contract research organisations are running greater numbers of clinical trials to bring life-saving treatments to patients. As trials increase in complexity and number, these organisations are under increased pressure to design and run faster, more efficient and more cost-effective studies.
CMDRs are fast becoming the tool of choice to help meet these challenges by providing the structure and tools to standardise and control metadata. By embedding CDISC standards in every part of their structure, CMDRs enable the creation of pre-approved, reusable metadata that meets regulatory standards from the outset. Furthermore, CMDRs power the automated tasks that reduce errors and time-consuming manual rework, helping teams build studies with efficiencies built-in as standard.
Creating a single source of truth for all trial metadata enables teams to access a central hub for collaboration, governance and visibility. This, in turn, leads to higher quality outputs that meet regulatory requirements, and facilitates optimised, cost-effective trials.
REFERENCES
1. clinicaltrials.gov/ct2/resources/trends, site visited on 27
September 2021.


